Explore Workflows
View already parsed workflows here or click here to add your own
| Graph | Name | Retrieved From | View |
|---|---|---|---|
|
|
cnv_exomedepth
CNV ExomeDepth calling |
Path: structuralvariants/cwl/subworkflows/cnv_exome_depth.cwl Branch/Commit ID: 1.0.7 |
|
|
|
trimmed_fastq
Quality Control (raw data), Raw Data trimming and Quality Control (pre-processed) |
Path: structuralvariants/cwl/subworkflows/trimmed_fastq.cwl Branch/Commit ID: 1.0.9 |
|
|
|
io-int-default-tool-and-wf.cwl
|
Path: tests/io-int-default-tool-and-wf.cwl Branch/Commit ID: main |
|
|
|
GATK-complete-WES-Workflow-h3abionet.cwl
# H3ABioNet GATK Germline Workflow # Overview A [GATK best-practices](https://software.broadinstitute.org/gatk/best-practices/bp_3step.php?case=GermShortWGS) germline workflow designed to work with GATK 3.5 (Van der Auwera et al., 2013). For more information see our [GitHub](https://github.com/h3abionet/h3agatk) site. # Workflow Summary 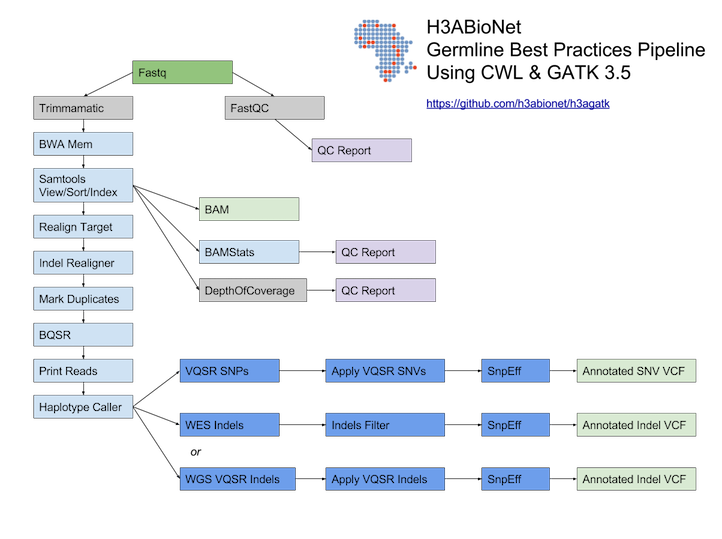 # Workflow Tool Details ## FastQC FastQC is used as an initial QC step where the input files are checked for usual metrics such as: - Read length - Reads distribution - GC content - ... ## Trimmomatic Trimmomatic is the entry point of the pipeline, it is used to cleanup the reads in the input fastq files from any sequencing adaptors. ## BWA [BWA](http://bio-bwa.sourceforge.net) is used to align the reads from the the input fastq files -paired-ends- (Li, 2013). We use specifically `bwa mem` as recommended by the [GATK best-practices](https://software.broadinstitute.org/gatk/best-practices/bp_3step.php?case=GermShortWGS). BWA produces a SAM file containing the aligned reads against the human reference genome (hg19, GATK bundle build 2.8). As GATK tools downstream requires properly formatted Read Group information. We add by default 'toy' Read Group information while processing the alignment to the output SAM file. we specifically use the flag `-R '@RG\tID:foo\tSM:bar\tLB:library1'`. ## SAMtools [SAMtools](http://www.htslib.org) (Li et al., 2009) are used few times in the pipeline: 1. Convert BWA's output from a SAM format to a BAM format 2. Sort the reads in the generated BAM file in step 1 (above) 3. Indexing the BAM file for the following tools to use ## Picard [Picard tools](https://broadinstitute.github.io/picard/) are used to mark duplicated reads in the aligned and sorted BAM file, making thus the files lighter and less prone to errors in the downstream steps of the pipeline. ## GATK [Genome Analysis Tool Kit](https://software.broadinstitute.org/gatk) refered to as GATK (DePristo et al., 2011) is used to process the data throught multiple steps as described by the [GATK best-practices](https://software.broadinstitute.org/gatk/best-practices/bp_3step.php?case=GermShortWGS) (i.e. figure bellow). 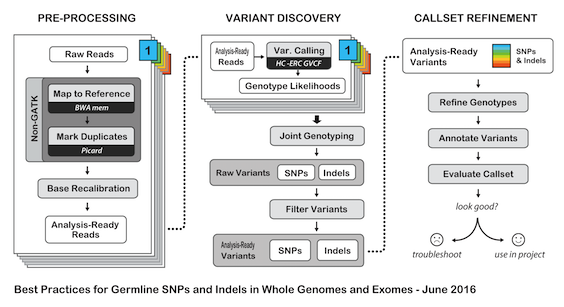 The GATK steps are the following: 1. Indel Realignment: 1. [Realign Target Creator](https://software.broadinstitute.org/gatk/documentation/tooldocs/org_broadinstitute_gatk_tools_walkers_indels_RealignerTargetCreator.php) 2. [Indel Realigner](https://software.broadinstitute.org/gatk/documentation/tooldocs/org_broadinstitute_gatk_tools_walkers_indels_IndelRealigner.php) 2. Mark Duplicates (a picard step) 3. Base Quality Score Recalibration (BQSR): 1. [Base Recalibrator](https://software.broadinstitute.org/gatk/documentation/tooldocs/org_broadinstitute_gatk_tools_walkers_bqsr_BaseRecalibrator.php) 2. [Print Reads](https://software.broadinstitute.org/gatk/documentation/tooldocs/org_broadinstitute_gatk_tools_walkers_readutils_PrintReads.php) 4. [Haplotype Caller](https://software.broadinstitute.org/gatk/documentation/tooldocs/) 5. Variant Quality Score Recalibration (VQSR): 1. [Variant Recalibrator](https://software.broadinstitute.org/gatk/documentation/tooldocs/org_broadinstitute_gatk_tools_walkers_variantrecalibration_VariantRecalibrator.php) 2. [Apply Recalibration](https://software.broadinstitute.org/gatk/documentation/tooldocs/org_broadinstitute_gatk_tools_walkers_variantrecalibration_ApplyRecalibration.php) ## SnpEff SNPEff is used in this pipeline to annotate the variant calls (Cingolani et al., 2012). The annotation is extensive and uses multi-database approach to provide the user with as much information about the called variants as possible. ## BAMStat [BAMStats](http://bamstats.sourceforge.net), is a simple software tool built on the Picard Java API (2), which can calculate and graphically display various metrics derived from SAM/BAM files of value in QC assessments. |
Path: workflows/GATK/GATK-complete-WES-Workflow-h3abionet.cwl Branch/Commit ID: cwl_v1_0 |
|
|
|
WF5201.cwl
|
Path: WF5201.cwl Branch/Commit ID: main |
|
|
|
wf_split_self_and_idr.cwl
This workflow returns the reproducible number of split peaks given a single bam file and its size-matched input pair. This workflow splits the bam file first, but does not do anything to the input. |
Path: cwl/wf_split_self_and_idr.cwl Branch/Commit ID: master |
|
|
|
hipepipe.cwl
|
Path: CWL-pipeline/hipepipe.cwl Branch/Commit ID: master |
|
|
|
iwdr_with_nested_dirs.cwl
|
Path: tests/iwdr_with_nested_dirs.cwl Branch/Commit ID: master |
|
|
|
protein annotation
Proteins - predict, cluster, identify, annotate |
Path: CWL/Workflows/protein-annotation.workflow.cwl Branch/Commit ID: master |
|
|
|
wf-loadContents3.cwl
|
Path: tests/wf-loadContents3.cwl Branch/Commit ID: main |

