Explore Workflows
View already parsed workflows here or click here to add your own
| Graph | Name | Retrieved From | View |
|---|---|---|---|
|
|
RNA-Seq pipeline paired-end
The original [BioWardrobe's](https://biowardrobe.com) [PubMed ID:26248465](https://www.ncbi.nlm.nih.gov/pubmed/26248465) **RNA-Seq** basic analysis for a **paired-end** experiment. A corresponded input [FASTQ](http://maq.sourceforge.net/fastq.shtml) file has to be provided. Current workflow should be used only with the paired-end RNA-Seq data. It performs the following steps: 1. Use STAR to align reads from input FASTQ files according to the predefined reference indices; generate unsorted BAM file and alignment statistics file 2. Use fastx_quality_stats to analyze input FASTQ files and generate quality statistics files 3. Use samtools sort to generate coordinate sorted BAM(+BAI) file pair from the unsorted BAM file obtained on the step 1 (after running STAR) 4. Generate BigWig file on the base of sorted BAM file 5. Map input FASTQ files to predefined rRNA reference indices using Bowtie to define the level of rRNA contamination; export resulted statistics to file 6. Calculate isoform expression level for the sorted BAM file and GTF/TAB annotation file using GEEP reads-counting utility; export results to file |
Path: workflows/rnaseq-pe.cwl Branch/Commit ID: 92f1a6da9c4f85fb51340b01b32373a50fde0891 |
|
|
|
RNA-Seq pipeline single-read
The original [BioWardrobe's](https://biowardrobe.com) [PubMed ID:26248465](https://www.ncbi.nlm.nih.gov/pubmed/26248465) **RNA-Seq** basic analysis for a **single-read** experiment. A corresponded input [FASTQ](http://maq.sourceforge.net/fastq.shtml) file has to be provided. Current workflow should be used only with the single-read RNA-Seq data. It performs the following steps: 1. Use STAR to align reads from input FASTQ file according to the predefined reference indices; generate unsorted BAM file and alignment statistics file 2. Use fastx_quality_stats to analyze input FASTQ file and generate quality statistics file 3. Use samtools sort to generate coordinate sorted BAM(+BAI) file pair from the unsorted BAM file obtained on the step 1 (after running STAR) 5. Generate BigWig file on the base of sorted BAM file 6. Map input FASTQ file to predefined rRNA reference indices using Bowtie to define the level of rRNA contamination; export resulted statistics to file 7. Calculate isoform expression level for the sorted BAM file and GTF/TAB annotation file using GEEP reads-counting utility; export results to file |
Path: workflows/rnaseq-se.cwl Branch/Commit ID: 92f1a6da9c4f85fb51340b01b32373a50fde0891 |
|
|
|
ChIP-Seq pipeline single-read
# ChIP-Seq basic analysis workflow for single-read data Reads are aligned to the reference genome with [Bowtie](http://bowtie-bio.sourceforge.net/index.shtml). Results are saved as coordinate sorted [BAM](http://samtools.github.io/hts-specs/SAMv1.pdf) alignment and index BAI files. Optionally, PCR duplicates can be removed. To obtain coverage in [bigWig](https://genome.ucsc.edu/goldenpath/help/bigWig.html) format, average fragment length is calculated by [MACS2](https://github.com/taoliu/MACS), and individual reads are extended to this length in the 3’ direction. Areas of enrichment identified by MACS2 are saved in ENCODE [narrow peak](http://genome.ucsc.edu/FAQ/FAQformat.html#format12) or [broad peak](https://genome.ucsc.edu/FAQ/FAQformat.html#format13) formats. Called peaks together with the nearest genes are saved in TSV format. In addition to basic statistics (number of total/mapped/multi-mapped/unmapped/duplicate reads), pipeline generates several quality control measures. Base frequency plots are used to estimate adapter contamination, a frequent occurrence in low-input ChIP-Seq experiments. Expected distinct reads count from [Preseq](http://smithlabresearch.org/software/preseq/) can be used to estimate read redundancy for a given sequencing depth. Average tag density profiles can be used to estimate ChIP enrichment for promoter proximal histone modifications. Use of different parameters for different antibodies (calling broad or narrow peaks) is possible. Additionally, users can elect to use BAM file from another experiment as control for MACS2 peak calling. ## Cite as *Kartashov AV, Barski A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 2015;16(1):158. Published 2015 Aug 7. [doi:10.1186/s13059-015-0720-3](https://www.ncbi.nlm.nih.gov/pubmed/26248465)* ## Software versions - Bowtie 1.2.0 - Samtools 1.4 - Preseq 2.0 - MACS2 2.1.1.20160309 - Bedtools 2.26.0 - UCSC userApps v358 ## Inputs | ID | Label | Description | Required | Default | Upstream analyses | | ------------------------- | ---------------------------------------------- | ---------------------------------------------------------------------------------------------------------------------------------------------------------------- | :------: | ------- | ------------------------------- | | **fastq\_file** | FASTQ file | Single-read sequencing data in FASTQ format (fastq, fq, bzip2, gzip, zip) | + | | | | **indices\_folder** | Genome indices | Directory with the genome indices generated by Bowtie | + | | genome\_indices/bowtie\_indices | | **annotation\_file** | Genome annotation file | Genome annotation file in TSV format | + | | genome\_indices/annotation | | **genome\_size** | Effective genome size | The length of the mappable genome (hs, mm, ce, dm or number, for example 2.7e9) | + | | genome\_indices/genome\_size | | **chrom\_length** | Chromosome lengths file | Chromosome lengths file in TSV format | + | | genome\_indices/chrom\_length | | **broad\_peak** | Call broad peaks | Make MACS2 call broad peaks by linking nearby highly enriched regions | + | | | | **control\_file** | Control ChIP-Seq single-read experiment | Indexed BAM file from the ChIP-Seq single-read experiment to be used as a control for MACS2 peak calling | | Null | control\_file/bambai\_pair | | **exp\_fragment\_size** | Expected fragment size | Expected fragment size for read extenstion towards 3' end if *force\_fragment\_size* was set to True or if calculated by MACS2 fragment size was less that 80 bp | | 150 | | | **force\_fragment\_size** | Force peak calling with expected fragment size | Make MACS2 don't build the shifting model and use expected fragment size for read extenstion towards 3' end | | False | | | **clip\_3p\_end** | Clip from 3' end | Number of base pairs to clip from 3' end | | 0 | | | **clip\_5p\_end** | Clip from 5' end | Number of base pairs to clip from 5' end | | 0 | | | **remove\_duplicates** | Remove PCR duplicates | Remove PCR duplicates from sorted BAM file | | False | | | **threads** | Number of threads | Number of threads for those steps that support multithreading | | 2 | | ## Outputs | ID | Label | Description | Required | Visualization | | ------------------------ | ---------------------------------- | ------------------------------------------------------------------------------------ | :------: | ------------------------------------------------------------------ | | **fastx\_statistics** | FASTQ quality statistics | FASTQ quality statistics in TSV format | + | *Base Frequency* and *Quality Control* plots in *QC Plots* tab | | **bambai\_pair** | Aligned reads | Coordinate sorted BAM alignment and index BAI files | + | *Nucleotide Sequence Alignments* track in *IGV Genome Browser* tab | | **bigwig** | Genome coverage | Genome coverage in bigWig format | + | *Genome Coverage* track in *IGV Genome Browser* tab | | **iaintersect\_result** | Gene annotated peaks | MACS2 peak file annotated with nearby genes | + | *Peak Coordinates* table in *Peak Calling* tab | | **atdp\_result** | Average Tag Density Plot | Average Tag Density Plot file in TSV format | + | *Average Tag Density Plot* in *QC Plots* tab | | **macs2\_called\_peaks** | Called peaks | Called peaks file with 1-based coordinates in XLS format | + | | | **macs2\_narrow\_peaks** | Narrow peaks | Called peaks file in ENCODE narrow peak format | | *Narrow peaks* track in *IGV Genome Browser* tab | | **macs2\_broad\_peaks** | Broad peaks | Called peaks file in ENCODE broad peak format | | *Broad peaks* track in *IGV Genome Browser* tab | | **preseq\_estimates** | Expected Distinct Reads Count Plot | Expected distinct reads count file from Preseq in TSV format | | *Expected Distinct Reads Count Plot* in *QC Plots* tab | | **workflow\_statistics** | Workflow execution statistics | Overall workflow execution statistics from bowtie\_aligner and samtools\_rmdup steps | + | *Overview* tab and experiment's preview | | **bowtie\_log** | Read alignment log | Read alignment log file from Bowtie | + | | |
Path: workflows/chipseq-se.cwl Branch/Commit ID: 92f1a6da9c4f85fb51340b01b32373a50fde0891 |
|
|
|
Build STAR indices
Workflow runs [STAR](https://github.com/alexdobin/STAR) v2.5.3a (03/17/2017) PMID: [23104886](https://www.ncbi.nlm.nih.gov/pubmed/23104886) to build indices for reference genome provided in a single FASTA file as fasta_file input and GTF annotation file from annotation_gtf_file input. Generated indices are saved in a folder with the name that corresponds to the input genome. |
Path: workflows/star-index.cwl Branch/Commit ID: 92f1a6da9c4f85fb51340b01b32373a50fde0891 |
|
|
|
Build Bismark indices
Copy fasta_file file to the folder and run run bismark_genome_preparation script to prepare indices for Bismark Methylation Analysis. Bowtie2 aligner is used by default. The name of the output indices folder is equal to the genome input. |
Path: workflows/bismark-index.cwl Branch/Commit ID: 92f1a6da9c4f85fb51340b01b32373a50fde0891 |
|
|
|
QuantSeq 3' mRNA-Seq single-read
### Pipeline for Lexogen's QuantSeq 3' mRNA-Seq Library Prep Kit FWD for Illumina [Lexogen original documentation](https://www.lexogen.com/quantseq-3mrna-sequencing/) * Cost-saving and streamlined globin mRNA depletion during QuantSeq library preparation * Genome-wide analysis of gene expression * Cost-efficient alternative to microarrays and standard RNA-Seq * Down to 100 pg total RNA input * Applicable for low quality and FFPE samples * Single-read sequencing of up to 9,216 samples/lane * Dual indexing and Unique Molecular Identifiers (UMIs) are available ### QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina The QuantSeq FWD Kit is a library preparation protocol designed to generate Illumina compatible libraries of sequences close to the 3’ end of polyadenylated RNA. QuantSeq FWD contains the Illumina Read 1 linker sequence in the second strand synthesis primer, hence NGS reads are generated towards the poly(A) tail, directly reflecting the mRNA sequence (see workflow). This version is the recommended standard for gene expression analysis. Lexogen furthermore provides a high-throughput version with optional dual indexing (i5 and i7 indices) allowing up to 9,216 samples to be multiplexed in one lane. #### Analysis of Low Input and Low Quality Samples The required input amount of total RNA is as low as 100 pg. QuantSeq is suitable to reproducibly generate libraries from low quality RNA, including FFPE samples. See Fig.1 and 2 for a comparison of two different RNA qualities (FFPE and fresh frozen cryo-block) of the same sample. 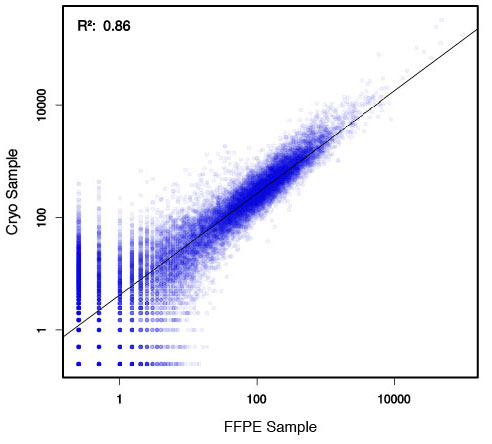 Figure 1 | Correlation of gene counts of FFPE and cryo samples. 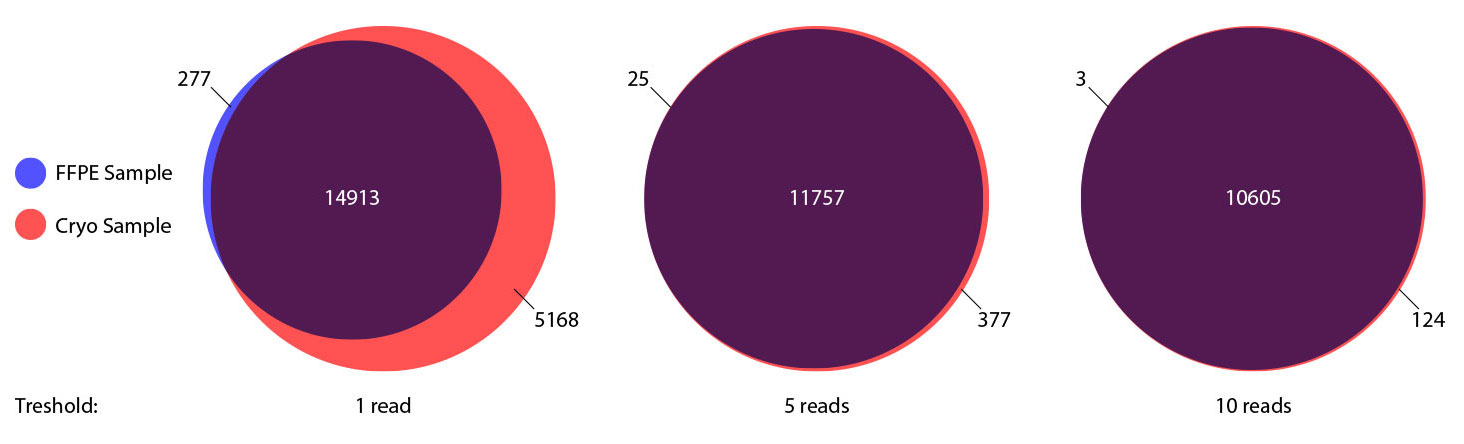 Figure 2 | Venn diagrams of genes detected by QuantSeq at a uniform read depth of 2.5 M reads in FFPE and cryo samples with 1, 5, and 10 reads/gene thresholds. #### Mapping of Transcript End Sites By using longer reads QuantSeq FWD allows to exactly pinpoint the 3’ end of poly(A) RNA (see Fig. 3) and therefore obtain accurate information about the 3’ UTR. 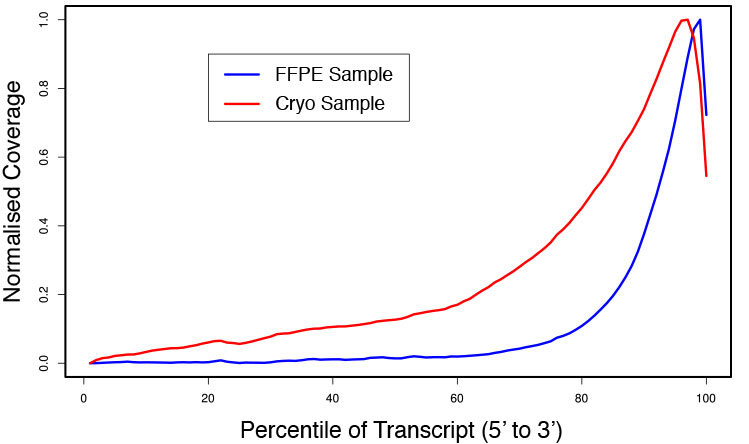 Figure 3 | QuantSeq read coverage versus normalized transcript length of NGS libraries derived from FFPE-RNA (blue) and cryo-preserved RNA (red). ### Current workflow should be used only with the single-end RNA-Seq data. It performs the following steps: 1. Separates UMIes and trims adapters from input FASTQ file 2. Uses ```STAR``` to align reads from input FASTQ file according to the predefined reference indices; generates unsorted BAM file and alignment statistics file 3. Uses ```fastx_quality_stats``` to analyze input FASTQ file and generates quality statistics file 4. Uses ```samtools sort``` and generates coordinate sorted BAM(+BAI) file pair from the unsorted BAM file obtained on the step 2 (after running STAR) 5. Uses ```umi_tools dedup``` and generates final filtered sorted BAM(+BAI) file pair 6. Generates BigWig file on the base of sorted BAM file 7. Maps input FASTQ file to predefined rRNA reference indices using ```bowtie``` to define the level of rRNA contamination; exports resulted statistics to file 8. Calculates isoform expression level for the sorted BAM file and GTF/TAB annotation file using GEEP reads-counting utility; exports results to file |
Path: workflows/trim-quantseq-mrnaseq-se.cwl Branch/Commit ID: 8049a781ac4aae579fbd3036fa0bf654532f15be |
|
|
|
Trim Galore SMARTer RNA-Seq pipeline paired-end strand specific
https://chipster.csc.fi/manual/library-type-summary.html Modified original [BioWardrobe's](https://biowardrobe.com) [PubMed ID:26248465](https://www.ncbi.nlm.nih.gov/pubmed/26248465) **RNA-Seq** basic analysis for a **pair-end** experiment. A corresponded input [FASTQ](http://maq.sourceforge.net/fastq.shtml) file has to be provided. Current workflow should be used only with the single-end RNA-Seq data. It performs the following steps: 1. Trim adapters from input FASTQ files 2. Use STAR to align reads from input FASTQ files according to the predefined reference indices; generate unsorted BAM file and alignment statistics file 3. Use fastx_quality_stats to analyze input FASTQ files and generate quality statistics files 4. Use samtools sort to generate coordinate sorted BAM(+BAI) file pair from the unsorted BAM file obtained on the step 1 (after running STAR) 5. Generate BigWig file on the base of sorted BAM file 6. Map input FASTQ files to predefined rRNA reference indices using Bowtie to define the level of rRNA contamination; export resulted statistics to file 7. Calculate isoform expression level for the sorted BAM file and GTF/TAB annotation file using GEEP reads-counting utility; export results to file |
Path: workflows/trim-rnaseq-pe-smarter-dutp.cwl Branch/Commit ID: 8a92669a566589d80fde9d151054ffc220ed4ddd |
|
|
|
CLIP-Seq pipeline for single-read experiment NNNNG
Cross-Linking ImmunoPrecipitation ================================= `CLIP` (`cross-linking immunoprecipitation`) is a method used in molecular biology that combines UV cross-linking with immunoprecipitation in order to analyse protein interactions with RNA or to precisely locate RNA modifications (e.g. m6A). (Uhl|Houwaart|Corrado|Wright|Backofen|2017)(Ule|Jensen|Ruggiu|Mele|2003)(Sugimoto|König|Hussain|Zupan|2012)(Zhang|Darnell|2011) (Ke| Alemu| Mertens| Gantman|2015) CLIP-based techniques can be used to map RNA binding protein binding sites or RNA modification sites (Ke| Alemu| Mertens| Gantman|2015)(Ke| Pandya-Jones| Saito| Fak|2017) of interest on a genome-wide scale, thereby increasing the understanding of post-transcriptional regulatory networks. The identification of sites where RNA-binding proteins (RNABPs) interact with target RNAs opens the door to understanding the vast complexity of RNA regulation. UV cross-linking and immunoprecipitation (CLIP) is a transformative technology in which RNAs purified from _in vivo_ cross-linked RNA-protein complexes are sequenced to reveal footprints of RNABP:RNA contacts. CLIP combined with high-throughput sequencing (HITS-CLIP) is a generalizable strategy to produce transcriptome-wide maps of RNA binding with higher accuracy and resolution than standard RNA immunoprecipitation (RIP) profiling or purely computational approaches. The application of CLIP to Argonaute proteins has expanded the utility of this approach to mapping binding sites for microRNAs and other small regulatory RNAs. Finally, recent advances in data analysis take advantage of cross-link–induced mutation sites (CIMS) to refine RNA-binding maps to single-nucleotide resolution. Once IP conditions are established, HITS-CLIP takes ~8 d to prepare RNA for sequencing. Established pipelines for data analysis, including those for CIMS, take 3–4 d. Workflow -------- CLIP begins with the in-vivo cross-linking of RNA-protein complexes using ultraviolet light (UV). Upon UV exposure, covalent bonds are formed between proteins and nucleic acids that are in close proximity. (Darnell|2012) The cross-linked cells are then lysed, and the protein of interest is isolated via immunoprecipitation. In order to allow for sequence specific priming of reverse transcription, RNA adapters are ligated to the 3' ends, while radiolabeled phosphates are transferred to the 5' ends of the RNA fragments. The RNA-protein complexes are then separated from free RNA using gel electrophoresis and membrane transfer. Proteinase K digestion is then performed in order to remove protein from the RNA-protein complexes. This step leaves a peptide at the cross-link site, allowing for the identification of the cross-linked nucleotide. (König| McGlincy| Ule|2012) After ligating RNA linkers to the RNA 5' ends, cDNA is synthesized via RT-PCR. High-throughput sequencing is then used to generate reads containing distinct barcodes that identify the last cDNA nucleotide. Interaction sites can be identified by mapping the reads back to the transcriptome. |
Path: workflows/clipseq-se.cwl Branch/Commit ID: 8049a781ac4aae579fbd3036fa0bf654532f15be |
|
|
|
RNA-Seq pipeline single-read strand specific
Note: should be updated The original [BioWardrobe's](https://biowardrobe.com) [PubMed ID:26248465](https://www.ncbi.nlm.nih.gov/pubmed/26248465) **RNA-Seq** basic analysis for **strand specific single-read** experiment. A corresponded input [FASTQ](http://maq.sourceforge.net/fastq.shtml) file has to be provided. Current workflow should be used only with the single-read RNA-Seq data. It performs the following steps: 1. Use STAR to align reads from input FASTQ file according to the predefined reference indices; generate unsorted BAM file and alignment statistics file 2. Use fastx_quality_stats to analyze input FASTQ file and generate quality statistics file 3. Use samtools sort to generate coordinate sorted BAM(+BAI) file pair from the unsorted BAM file obtained on the step 1 (after running STAR) 5. Generate BigWig file on the base of sorted BAM file 6. Map input FASTQ file to predefined rRNA reference indices using Bowtie to define the level of rRNA contamination; export resulted statistics to file 7. Calculate isoform expression level for the sorted BAM file and GTF/TAB annotation file using GEEP reads-counting utility; export results to file |
Path: workflows/rnaseq-se-dutp.cwl Branch/Commit ID: 7fb8a1ebf8145791440bc2fed9c5f2d78a19d04c |
|
|
|
DESeq - differential gene expression analysis
Differential gene expression analysis ===================================== Differential gene expression analysis based on the negative binomial distribution Estimate variance-mean dependence in count data from high-throughput sequencing assays and test for differential expression based on a model using the negative binomial distribution. DESeq1 ------ High-throughput sequencing assays such as RNA-Seq, ChIP-Seq or barcode counting provide quantitative readouts in the form of count data. To infer differential signal in such data correctly and with good statistical power, estimation of data variability throughout the dynamic range and a suitable error model are required. Simon Anders and Wolfgang Huber propose a method based on the negative binomial distribution, with variance and mean linked by local regression and present an implementation, [DESeq](http://bioconductor.org/packages/release/bioc/html/DESeq.html), as an R/Bioconductor package DESeq2 ------ In comparative high-throughput sequencing assays, a fundamental task is the analysis of count data, such as read counts per gene in RNA-seq, for evidence of systematic changes across experimental conditions. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. [DESeq2](http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html), a method for differential analysis of count data, using shrinkage estimation for dispersions and fold changes to improve stability and interpretability of estimates. This enables a more quantitative analysis focused on the strength rather than the mere presence of differential expression. |
Path: workflows/deseq.cwl Branch/Commit ID: 7fb8a1ebf8145791440bc2fed9c5f2d78a19d04c |

