Explore Workflows
View already parsed workflows here or click here to add your own
| Graph | Name | Retrieved From | View |
|---|---|---|---|
|
|
bam to trimmed fastqs and biscuit alignments
|
Path: definitions/subworkflows/bam_to_trimmed_fastq_and_biscuit_alignments.cwl Branch/Commit ID: e649fcb1092905c539be026a3f23c82d5b0871d2 |
|
|
|
allele-process-strain.cwl
|
Path: subworkflows/allele-process-strain.cwl Branch/Commit ID: bfa3843bcf36125ff258d6314f64b41336f06e6b |
|
|
|
Trim Galore ChIP-Seq pipeline paired-end
This ChIP-Seq pipeline is based on the original [BioWardrobe's](https://biowardrobe.com) [PubMed ID:26248465](https://www.ncbi.nlm.nih.gov/pubmed/26248465) **ChIP-Seq** basic analysis workflow for a **paired-end** experiment with Trim Galore. ### Data Analysis SciDAP starts from the .fastq files which most DNA cores and commercial NGS companies return. Starting from raw data allows us to ensure that all experiments have been processed in the same way and simplifies the deposition of data to GEO upon publication. The data can be uploaded from users computer, downloaded directly from an ftp server of the core facility by providing a URL or from GEO by providing SRA accession number. Our current pipelines include the following steps: 1. Trimming the adapters with TrimGalore. This step is particularly important when the reads are long and the fragments are short-resulting in sequencing adapters at the end of read. If adapter is not removed the read will not map. TrimGalore can recognize standard adapters, such as Illumina or Nexterra/Tn5 adapters. 2. QC 3. (Optional) trimming adapters on 5' or 3' end by the specified number of bases. 4. Mapping reads with BowTie. Only uniquely mapped reads with less than 3 mismatches are used in the downstream analysis. Results are saved as a .bam file. 5. (Optional) Removal of duplicates (reads/pairs of reads mapping to exactly same location). This step is used to remove reads overamplified in PCR. Unfortunately, it may also remove \"good\" reads. We usually do not remove duplicates unless the library is heavily duplicated. Please note that MACS2 will remove 'excessive' duplicates during peak calling ina smart way (those not supported by other nearby reads). 6. Peakcalling by MACS2. (Optionally), it is possible to specify read extension length for MACS2 to use if the length determined automatically is wrong. 7. Generation of BigWig coverage files for display on the browser. The coverage shows the number of fragments at each base in the genome normalized to the number of millions of mapped reads. In the case of PE sequencing the fragments are real, but in the case of single reads the fragments are estimated by extending reads to the average fragment length found by MACS2 or specified by the user in 6. ### Details _Trim Galore_ is a wrapper around [Cutadapt](https://github.com/marcelm/cutadapt) and [FastQC](http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to consistently apply adapter and quality trimming to FastQ files, with extra functionality for RRBS data. A [FASTQ](http://maq.sourceforge.net/fastq.shtml) input file has to be provided. In outputs it returns coordinate sorted BAM file alongside with index BAI file, quality statistics for both the input FASTQ files, reads coverage in a form of BigWig file, peaks calling data in a form of narrowPeak or broadPeak files, islands with the assigned nearest genes and region type, data for average tag density plot (on the base of BAM file). Workflow starts with running fastx_quality_stats (steps fastx_quality_stats_upstream and fastx_quality_stats_downstream) from FASTX-Toolkit to calculate quality statistics for both upstream and downstream input FASTQ files. At the same time Bowtie is used to align reads from input FASTQ files to reference genome (Step bowtie_aligner). The output of this step is unsorted SAM file which is being sorted and indexed by samtools sort and samtools index (Step samtools_sort_index). Depending on workflow’s input parameters indexed and sorted BAM file could be processed by samtools rmdup (Step samtools_rmdup) to remove all possible read duplicates. In a case when removing duplicates is not necessary the step returns original input BAM and BAI files without any processing. If the duplicates were removed the following step (Step samtools_sort_index_after_rmdup) reruns samtools sort and samtools index with BAM and BAI files, if not - the step returns original unchanged input files. Right after that macs2 callpeak performs peak calling (Step macs2_callpeak). On the base of returned outputs the next step (Step macs2_island_count) calculates the number of islands and estimated fragment size. If the last one is less that 80 (hardcoded in a workflow) macs2 callpeak is rerun again with forced fixed fragment size value (Step macs2_callpeak_forced). If at the very beginning it was set in workflow input parameters to force run peak calling with fixed fragment size, this step is skipped and the original peak calling results are saved. In the next step workflow again calculates the number of islands and estimated fragment size (Step macs2_island_count_forced) for the data obtained from macs2_callpeak_forced step. If the last one was skipped the results from macs2_island_count_forced step are equal to the ones obtained from macs2_island_count step. Next step (Step macs2_stat) is used to define which of the islands and estimated fragment size should be used in workflow output: either from macs2_island_count step or from macs2_island_count_forced step. If input trigger of this step is set to True it means that macs2_callpeak_forced step was run and it returned different from macs2_callpeak step results, so macs2_stat step should return [fragments_new, fragments_old, islands_new], if trigger is False the step returns [fragments_old, fragments_old, islands_old], where sufix \"old\" defines results obtained from macs2_island_count step and sufix \"new\" - from macs2_island_count_forced step. The following two steps (Step bamtools_stats and bam_to_bigwig) are used to calculate coverage on the base of input BAM file and save it in BigWig format. For that purpose bamtools stats returns the number of mapped reads number which is then used as scaling factor by bedtools genomecov when it performs coverage calculation and saves it in BED format. The last one is then being sorted and converted to BigWig format by bedGraphToBigWig tool from UCSC utilities. Step get_stat is used to return a text file with statistics in a form of [TOTAL, ALIGNED, SUPRESSED, USED] reads count. Step island_intersect assigns genes and regions to the islands obtained from macs2_callpeak_forced. Step average_tag_density is used to calculate data for average tag density plot on the base of BAM file. |
Path: workflows/trim-chipseq-pe.cwl Branch/Commit ID: e0a30aa1ad516dd2ec0e9ce006428964b840daf4 |
|
|
|
allele-process-reference.cwl
|
Path: subworkflows/allele-process-reference.cwl Branch/Commit ID: bfa3843bcf36125ff258d6314f64b41336f06e6b |
|
|
|
running cellranger mkfastq and count
|
Path: definitions/subworkflows/cellranger_mkfastq_and_count.cwl Branch/Commit ID: e649fcb1092905c539be026a3f23c82d5b0871d2 |
|
|
|
genomics-workspace-transcript.cwl
|
Path: flow_genomicsWorkspace/genomics-workspace-transcript.cwl Branch/Commit ID: f7894707dd30a0edd199d3b67c4c8678f64c90b3 |
|
|
|
bacterial_orthology
|
Path: bacterial_orthology/wf_bacterial_orthology.cwl Branch/Commit ID: 22ffe27d9d4a899def7592d75d5871c1856adbdb |
|
|
|
genomics-workspace-genome.cwl
|
Path: flow_genomicsWorkspace/genomics-workspace-genome.cwl Branch/Commit ID: f7894707dd30a0edd199d3b67c4c8678f64c90b3 |
|
|
|
kmer_cache_retrieve
|
Path: task_types/tt_kmer_cache_retrieve.cwl Branch/Commit ID: 546742b523ce12f6246a52c838a51920a08dad4b |
|
|
|
QuantSeq 3' mRNA-Seq single-read
### Pipeline for Lexogen's QuantSeq 3' mRNA-Seq Library Prep Kit FWD for Illumina [Lexogen original documentation](https://www.lexogen.com/quantseq-3mrna-sequencing/) * Cost-saving and streamlined globin mRNA depletion during QuantSeq library preparation * Genome-wide analysis of gene expression * Cost-efficient alternative to microarrays and standard RNA-Seq * Down to 100 pg total RNA input * Applicable for low quality and FFPE samples * Single-read sequencing of up to 9,216 samples/lane * Dual indexing and Unique Molecular Identifiers (UMIs) are available ### QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina The QuantSeq FWD Kit is a library preparation protocol designed to generate Illumina compatible libraries of sequences close to the 3’ end of polyadenylated RNA. QuantSeq FWD contains the Illumina Read 1 linker sequence in the second strand synthesis primer, hence NGS reads are generated towards the poly(A) tail, directly reflecting the mRNA sequence (see workflow). This version is the recommended standard for gene expression analysis. Lexogen furthermore provides a high-throughput version with optional dual indexing (i5 and i7 indices) allowing up to 9,216 samples to be multiplexed in one lane. #### Analysis of Low Input and Low Quality Samples The required input amount of total RNA is as low as 100 pg. QuantSeq is suitable to reproducibly generate libraries from low quality RNA, including FFPE samples. See Fig.1 and 2 for a comparison of two different RNA qualities (FFPE and fresh frozen cryo-block) of the same sample. 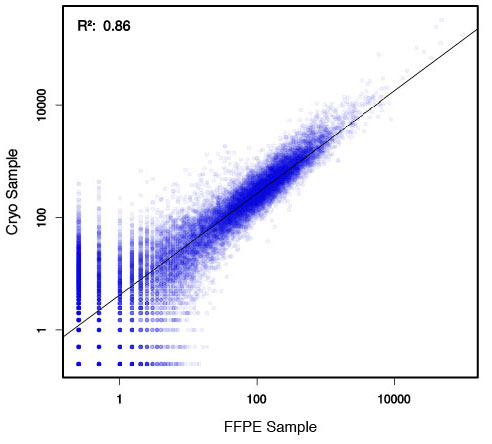 Figure 1 | Correlation of gene counts of FFPE and cryo samples. 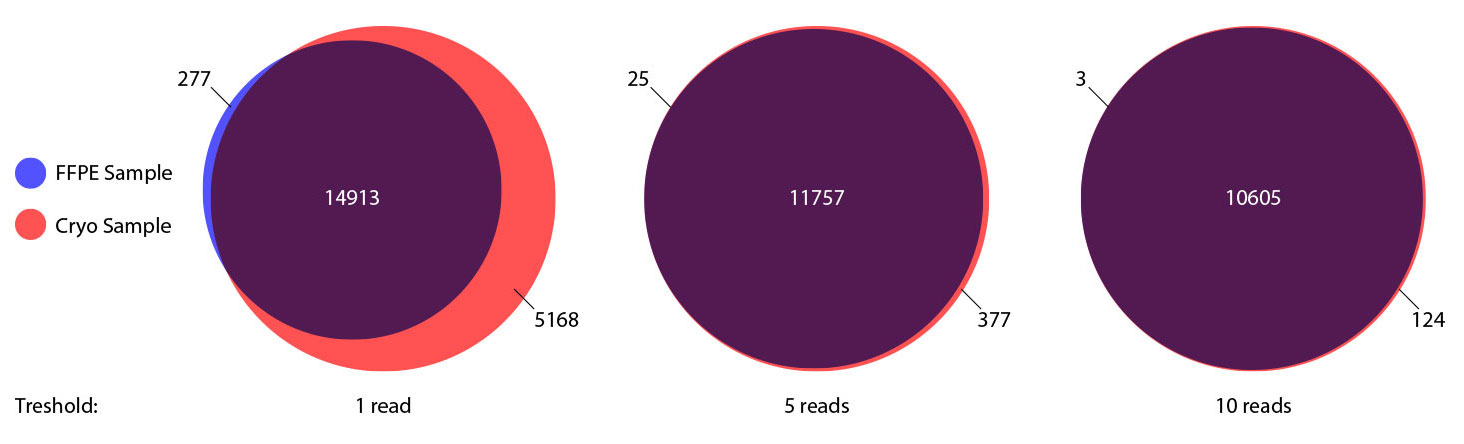 Figure 2 | Venn diagrams of genes detected by QuantSeq at a uniform read depth of 2.5 M reads in FFPE and cryo samples with 1, 5, and 10 reads/gene thresholds. #### Mapping of Transcript End Sites By using longer reads QuantSeq FWD allows to exactly pinpoint the 3’ end of poly(A) RNA (see Fig. 3) and therefore obtain accurate information about the 3’ UTR. 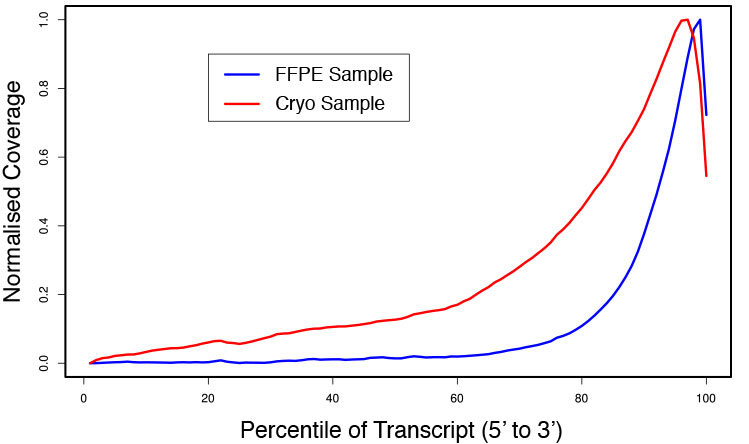 Figure 3 | QuantSeq read coverage versus normalized transcript length of NGS libraries derived from FFPE-RNA (blue) and cryo-preserved RNA (red). ### Current workflow should be used only with the single-end RNA-Seq data. It performs the following steps: 1. Separates UMIes and trims adapters from input FASTQ file 2. Uses ```STAR``` to align reads from input FASTQ file according to the predefined reference indices; generates unsorted BAM file and alignment statistics file 3. Uses ```fastx_quality_stats``` to analyze input FASTQ file and generates quality statistics file 4. Uses ```samtools sort``` and generates coordinate sorted BAM(+BAI) file pair from the unsorted BAM file obtained on the step 2 (after running STAR) 5. Uses ```umi_tools dedup``` and generates final filtered sorted BAM(+BAI) file pair 6. Generates BigWig file on the base of sorted BAM file 7. Maps input FASTQ file to predefined rRNA reference indices using ```bowtie``` to define the level of rRNA contamination; exports resulted statistics to file 8. Calculates isoform expression level for the sorted BAM file and GTF/TAB annotation file using GEEP reads-counting utility; exports results to file |
Path: workflows/trim-quantseq-mrnaseq-se.cwl Branch/Commit ID: 480e99a4bb3046e0565113d9dca294e0895d3b0c |

