Explore Workflows
View already parsed workflows here or click here to add your own
| Graph | Name | Retrieved From | View |
|---|---|---|---|
|
|
trim-rnaseq-se.cwl
Runs RNA-Seq BioWardrobe basic analysis with single-end data file. |
Path: workflows/trim-rnaseq-se.cwl Branch/Commit ID: 9a03dbe8829ca649814d9c8bd11fe3a673750a95 |
|
|
|
Subworkflow to allow calling cnvkit with cram instead of bam files
|
Path: definitions/subworkflows/cram_to_cnvkit.cwl Branch/Commit ID: c6bbd4cdd612b3b5cc6e9000df4800c21e192bf5 |
|
|
|
revsort.cwl
Reverse the lines in a document, then sort those lines. |
Path: tests/wf/revsort.cwl Branch/Commit ID: 63f539ba60e91f0cb3ce7cda2c5da5c65525c375 |
|
|
|
Running cellranger count and lineage inference
|
Path: definitions/subworkflows/single_cell_rnaseq.cwl Branch/Commit ID: 0805e8e0d358136468e0a9f49e06005e41965adc |
|
|
|
GSEApy - Gene Set Enrichment Analysis in Python
GSEAPY: Gene Set Enrichment Analysis in Python ============================================== Gene Set Enrichment Analysis is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (e.g. phenotypes). GSEA requires as input an expression dataset, which contains expression profiles for multiple samples. While the software supports multiple input file formats for these datasets, the tab-delimited GCT format is the most common. The first column of the GCT file contains feature identifiers (gene ids or symbols in the case of data derived from RNA-Seq experiments). The second column contains a description of the feature; this column is ignored by GSEA and may be filled with “NA”s. Subsequent columns contain the expression values for each feature, with one sample's expression value per column. It is important to note that there are no hard and fast rules regarding how a GCT file's expression values are derived. The important point is that they are comparable to one another across features within a sample and comparable to one another across samples. Tools such as DESeq2 can be made to produce properly normalized data (normalized counts) which are compatible with GSEA. Documents ============================================== - GSEA Home Page: https://www.gsea-msigdb.org/gsea/index.jsp - Results Interpretation: https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideTEXT.htm#_Interpreting_GSEA_Results - GSEA User Guide: https://gseapy.readthedocs.io/en/latest/faq.html - GSEAPY Docs: https://gseapy.readthedocs.io/en/latest/introduction.html References ============================================== - Subramanian, Tamayo, et al. (2005, PNAS), https://www.pnas.org/content/102/43/15545 - Mootha, Lindgren, et al. (2003, Nature Genetics), http://www.nature.com/ng/journal/v34/n3/abs/ng1180.html - Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 128(14). - Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 2016; gkw377 . - Xie Z, Bailey A, Kuleshov MV, Clarke DJB., Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, Jeon M, & Ma’ayan A. Gene set knowledge discovery with Enrichr. Current Protocols, 1, e90. 2021. doi: 10.1002/cpz1.90 |
Path: workflows/gseapy.cwl Branch/Commit ID: b4d578c2ba4713a5a22163d9f8c7105acda1f22e |
|
|
|
Align reference proteins plane complete workflow
|
Path: protein_alignment/wf_protein_alignment.cwl Branch/Commit ID: ce433f771ebf5677c9f40858e2ae91b1a7e75d30 |
|
|
|
struct_output.cwl
|
Path: wdl2cwl/tests/cwl_files/struct_output.cwl Branch/Commit ID: 81d4bdaecebaa843903b40834cb15e350aa047e8 |
|
|
|
CLIP-Seq pipeline for single-read experiment NNNNG
Cross-Linking ImmunoPrecipitation ================================= `CLIP` (`cross-linking immunoprecipitation`) is a method used in molecular biology that combines UV cross-linking with immunoprecipitation in order to analyse protein interactions with RNA or to precisely locate RNA modifications (e.g. m6A). (Uhl|Houwaart|Corrado|Wright|Backofen|2017)(Ule|Jensen|Ruggiu|Mele|2003)(Sugimoto|König|Hussain|Zupan|2012)(Zhang|Darnell|2011) (Ke| Alemu| Mertens| Gantman|2015) CLIP-based techniques can be used to map RNA binding protein binding sites or RNA modification sites (Ke| Alemu| Mertens| Gantman|2015)(Ke| Pandya-Jones| Saito| Fak|2017) of interest on a genome-wide scale, thereby increasing the understanding of post-transcriptional regulatory networks. The identification of sites where RNA-binding proteins (RNABPs) interact with target RNAs opens the door to understanding the vast complexity of RNA regulation. UV cross-linking and immunoprecipitation (CLIP) is a transformative technology in which RNAs purified from _in vivo_ cross-linked RNA-protein complexes are sequenced to reveal footprints of RNABP:RNA contacts. CLIP combined with high-throughput sequencing (HITS-CLIP) is a generalizable strategy to produce transcriptome-wide maps of RNA binding with higher accuracy and resolution than standard RNA immunoprecipitation (RIP) profiling or purely computational approaches. The application of CLIP to Argonaute proteins has expanded the utility of this approach to mapping binding sites for microRNAs and other small regulatory RNAs. Finally, recent advances in data analysis take advantage of cross-link–induced mutation sites (CIMS) to refine RNA-binding maps to single-nucleotide resolution. Once IP conditions are established, HITS-CLIP takes ~8 d to prepare RNA for sequencing. Established pipelines for data analysis, including those for CIMS, take 3–4 d. Workflow -------- CLIP begins with the in-vivo cross-linking of RNA-protein complexes using ultraviolet light (UV). Upon UV exposure, covalent bonds are formed between proteins and nucleic acids that are in close proximity. (Darnell|2012) The cross-linked cells are then lysed, and the protein of interest is isolated via immunoprecipitation. In order to allow for sequence specific priming of reverse transcription, RNA adapters are ligated to the 3' ends, while radiolabeled phosphates are transferred to the 5' ends of the RNA fragments. The RNA-protein complexes are then separated from free RNA using gel electrophoresis and membrane transfer. Proteinase K digestion is then performed in order to remove protein from the RNA-protein complexes. This step leaves a peptide at the cross-link site, allowing for the identification of the cross-linked nucleotide. (König| McGlincy| Ule|2012) After ligating RNA linkers to the RNA 5' ends, cDNA is synthesized via RT-PCR. High-throughput sequencing is then used to generate reads containing distinct barcodes that identify the last cDNA nucleotide. Interaction sites can be identified by mapping the reads back to the transcriptome. |
Path: workflows/clipseq-se.cwl Branch/Commit ID: 22880e0f41d0420a17d643e8a6e8ee18165bbfbf |
|
|
|
Single-Cell Preprocessing Cell Ranger Pipeline
Devel version of Single-Cell Preprocessing Cell Ranger Pipeline =============================================================== |
Path: workflows/single-cell-preprocess-cellranger.cwl Branch/Commit ID: 954bb2f213d97dfef1cddaf9e830169a92ad0c6b |
|
|
|
QuantSeq 3' mRNA-Seq single-read
### Pipeline for Lexogen's QuantSeq 3' mRNA-Seq Library Prep Kit FWD for Illumina [Lexogen original documentation](https://www.lexogen.com/quantseq-3mrna-sequencing/) * Cost-saving and streamlined globin mRNA depletion during QuantSeq library preparation * Genome-wide analysis of gene expression * Cost-efficient alternative to microarrays and standard RNA-Seq * Down to 100 pg total RNA input * Applicable for low quality and FFPE samples * Single-read sequencing of up to 9,216 samples/lane * Dual indexing and Unique Molecular Identifiers (UMIs) are available ### QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina The QuantSeq FWD Kit is a library preparation protocol designed to generate Illumina compatible libraries of sequences close to the 3’ end of polyadenylated RNA. QuantSeq FWD contains the Illumina Read 1 linker sequence in the second strand synthesis primer, hence NGS reads are generated towards the poly(A) tail, directly reflecting the mRNA sequence (see workflow). This version is the recommended standard for gene expression analysis. Lexogen furthermore provides a high-throughput version with optional dual indexing (i5 and i7 indices) allowing up to 9,216 samples to be multiplexed in one lane. #### Analysis of Low Input and Low Quality Samples The required input amount of total RNA is as low as 100 pg. QuantSeq is suitable to reproducibly generate libraries from low quality RNA, including FFPE samples. See Fig.1 and 2 for a comparison of two different RNA qualities (FFPE and fresh frozen cryo-block) of the same sample. 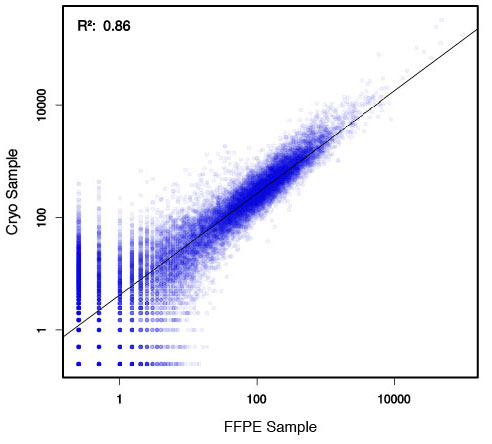 Figure 1 | Correlation of gene counts of FFPE and cryo samples. 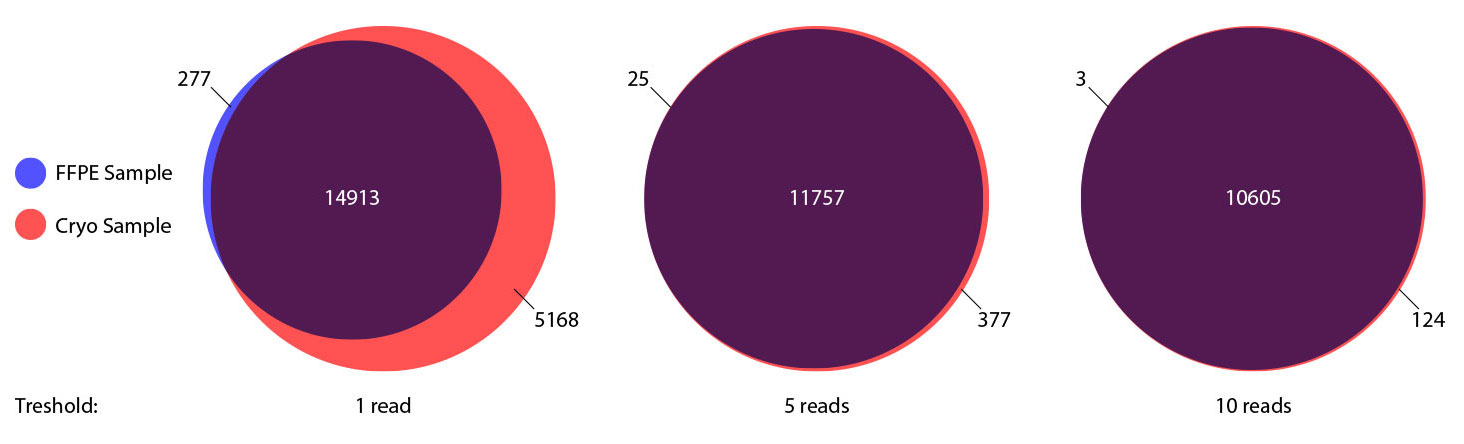 Figure 2 | Venn diagrams of genes detected by QuantSeq at a uniform read depth of 2.5 M reads in FFPE and cryo samples with 1, 5, and 10 reads/gene thresholds. #### Mapping of Transcript End Sites By using longer reads QuantSeq FWD allows to exactly pinpoint the 3’ end of poly(A) RNA (see Fig. 3) and therefore obtain accurate information about the 3’ UTR. 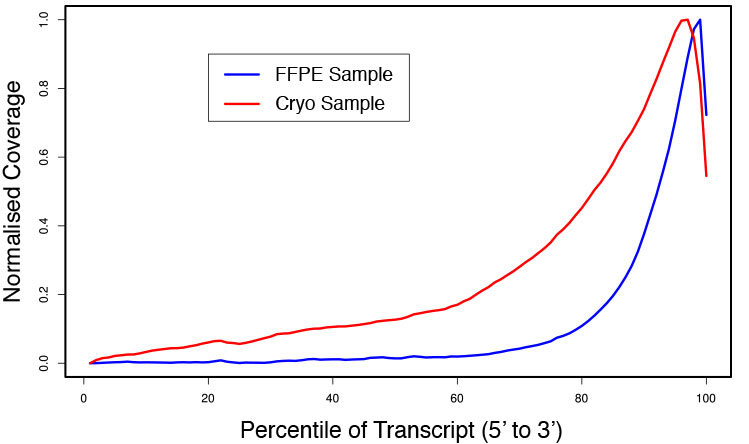 Figure 3 | QuantSeq read coverage versus normalized transcript length of NGS libraries derived from FFPE-RNA (blue) and cryo-preserved RNA (red). ### Current workflow should be used only with the single-end RNA-Seq data. It performs the following steps: 1. Separates UMIes and trims adapters from input FASTQ file 2. Uses ```STAR``` to align reads from input FASTQ file according to the predefined reference indices; generates unsorted BAM file and alignment statistics file 3. Uses ```fastx_quality_stats``` to analyze input FASTQ file and generates quality statistics file 4. Uses ```samtools sort``` and generates coordinate sorted BAM(+BAI) file pair from the unsorted BAM file obtained on the step 2 (after running STAR) 5. Uses ```umi_tools dedup``` and generates final filtered sorted BAM(+BAI) file pair 6. Generates BigWig file on the base of sorted BAM file 7. Maps input FASTQ file to predefined rRNA reference indices using ```bowtie``` to define the level of rRNA contamination; exports resulted statistics to file 8. Calculates isoform expression level for the sorted BAM file and GTF/TAB annotation file using GEEP reads-counting utility; exports results to file |
Path: workflows/trim-quantseq-mrnaseq-se.cwl Branch/Commit ID: 954bb2f213d97dfef1cddaf9e830169a92ad0c6b |

